遗传变异图谱是研究人群演化史、医学遗传学、基因型-表型关联的基础。此前,大多数全基因组测序相关研究主要集中在欧洲血统人群。已有研究表明,罕见和低频的变异往往是特定于人群或样本的,尤其是许多与疾病相关的变异。针对特定人群的基因组数据可以为全基因组关联研究、区域适应性研究、用药指导等提供更准确的参考。
单倍型参考面板可基于大型人群队列中已知的单倍型信息,对来源于相对稀疏的基因变异芯片或低覆盖率测序的样本中缺失的基因型进行推演,是促进全基因组关联研究 (genome-wide association study, GWAS) 的有意义且具有成本效益的方法。此前缺乏中国人群特异的参考面板,其他参考面板对中国人群特异的变异推演效果较差,从而导致GWAS中遗漏潜在的表型相关变异。
中国人群全基因组测序资源和单倍型参考面板的缺乏极大地阻碍了世界上最大人群的遗传学与精准医学研究。为此,金沙集团1862cc徐涛院士团队和何顺民研究员团队合作在国际学术期刊《Cell Reports》在线发表了题为"NyuWa Genome Resource: A Deep Whole Genome Sequencing-Based Variation Profile and Reference Panel for the Chinese Population"的文章(图1),介绍该团队关于"女娲"(NyuWa)中国人群基因组资源库(http://bigdata.ibp.ac.cn/NyuWa/)的工作,提供针对中国人群的遗传变异图谱与参考面板基因型推演服务,旨在促进中国人群的遗传学与医学研究。

图1. 文章发表于Cell Reports。
研究团队分析了 2,999 个中国人的全基因组深度测序数据(26.2X),并以中国神话中创造人类的女娲命名。基于NyuWa数据资源,构建了包含7106万SNPs 和819万 InDels的中国人群遗传变异图谱(图2),并对其进行全面注释。相比其它人群队列,NyuWa数据集包含2501万新的变异,其中包括14.9万非同义变异、10.1万有害变异、11493个编码和非编码基因的功能丧失变异、636个癌症相关基因的蛋白截短变异。大量的新变异表明,在以往的遗传研究中,中国人群的变异代表性不足,NyuWa基因组资源则填补了这一空缺。
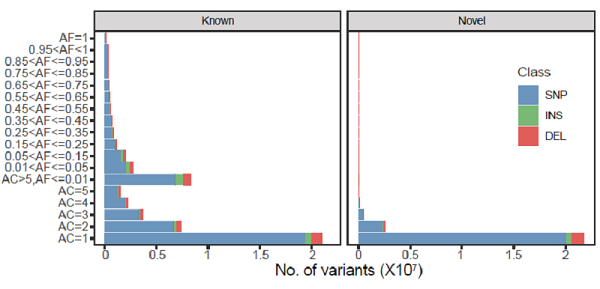
图2. NyuWa全基因组测序资源的变异数量。
此外根据临床相关数据库的注释,在NyuWa中发现了1,140个致病变异,以及药物基因组学相关位点(图3A)和癌症风险位点(图3B)上中国人群与世界其他人群的变异频率差异。这些发现有助于中国人群精准医学研究,可能促进新的遗传学和医学进展。

图3. NyuWa中的药物基因组学位点(A)与癌症风险基因座(B)变异。
汉族人口是东亚乃至全世界最大的民族,约占全球人口的 20%,为汉族人群构建一个完整的、大队列的、高质量的参考面板,对汉族的遗传学与医学研究具有重要参考价值。基于NyuWa数据资源,研究团队构建了包含 5804 个单倍型和 1926万 变异的单倍型参考面板,其中325万变异未包含在其它参考面板中,这些 NyuWa 参考面板特有的变异可能会在未来的关联研究中带来新的发现。这是第一个数千人级别公开可用的中国人群特异的单倍型参考面板。
为了评估NyuWa参考面板的基因型推演性能,研究团队使用来自人类基因组多样性计划(the Human Genome Diversity Project, HGDP)的亚洲各个人群芯片基因分型数据和高覆盖率 WGS 数据作为测试数据集。与其他参考面板相比,NyuWa 参考面板将汉族人群基因型推演的错误率降低了 30%-51%,在大多数其他东亚和东北亚人群中也有优异的表现(图4A-D)。研究团队进一步比较了不同等位基因频率的推算结果和实际基因型之间的相关性,NyuWa参考面板的性能在汉族的所有等位基因频率区间中均具有绝对优势(图4E)。此外,NyuWa参考面板与千人基因组(1KGP3)面板的结合进一步提高了亚洲人群的基因型推演效果。
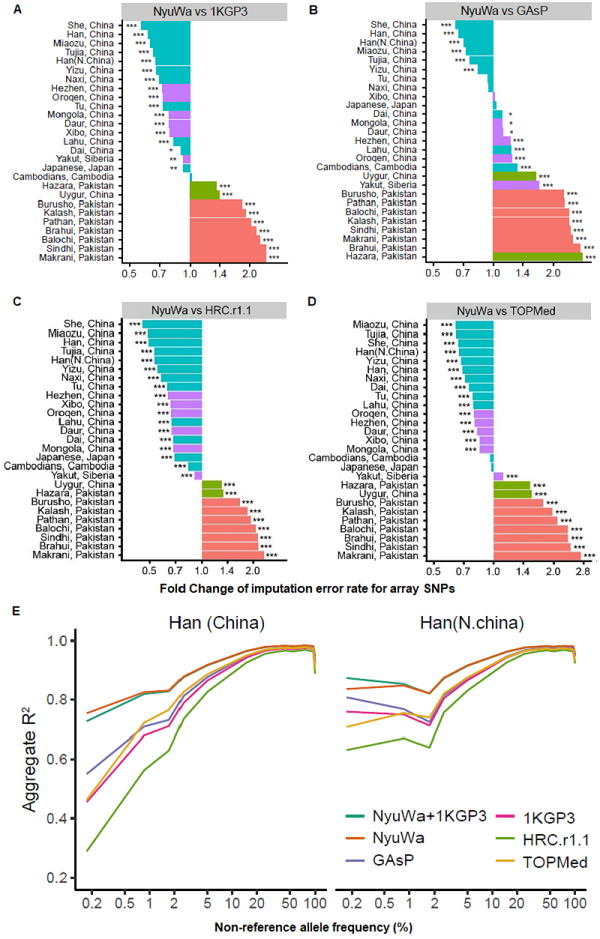
图4. NyuWa参考面板对汉族基因型推演具有最佳性能。
考虑到南北方汉族遗传差异的存在,研究团队将 NyuWa 参考面板中的样本分为北方和南方子集,使用子集样本分别构建北方和南方汉族的参考面板,通过基因型推演的模拟测试,证明以NyuWa的人群规模,一个南北整合的参考面板对中国北方人和南方人都适用(图5)。
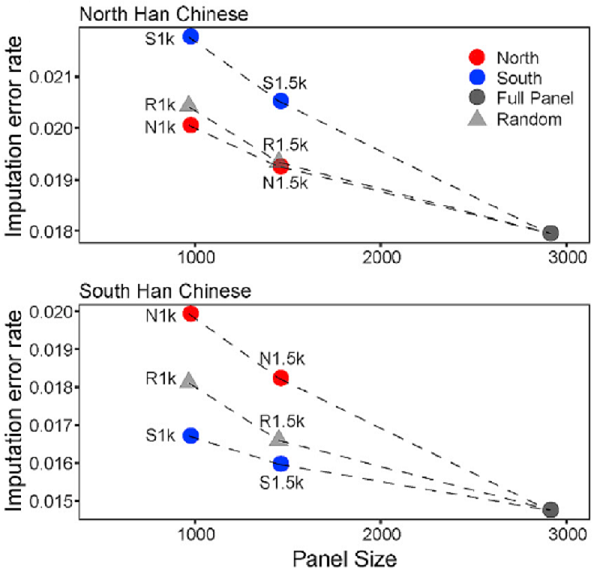
图5. 南北方汉族测试数据集的基因型推演错误率。
综上,基于中国人群的大型队列深度 WGS 数据,研究团队构建了中国人群的遗传变异图谱和首个数千人级别公开可用的中国人群单倍型参考面板,将所有结果整合为中国人群基因组资源库NyuWa(图6),旨在促进中国和亚洲人群的遗传学和精准医学研究。

图6. NyuWa资源库。
当前有关医学基因组学的知识和指南主要来自以欧洲人群为主的遗传和基因组资源,可能会遗漏有关非欧洲人群的遗传信息。亚洲人群起源、迁徙和融合历史悠久而复杂,使得其遗传多样性研究充满了挑战和机遇。作为世界上人口最多的国家,针对中国人群的全基因组测序工作,对于扩充世界人群遗传资源多样性、提高中国人群医学研究准确性非常必要,有助于深入了解亚洲人群结构与人群历史,并对寻找复杂疾病遗传因素的研究设计以及人口健康指导具有重要参考价值。
金沙集团1862cc的何顺民研究员、徐涛院士为该文共同通讯作者,金沙集团1862cc的副研究员张鹏博士、副研究员罗华夏博士、特别研究助理李燕燕博士、副研究员王友博士、博士研究生王佳佳、博士研究生郑宇为该文并列第一作者。本研究得到了金沙集团1862cc战略性先导科技专项、 国家自然科学基金、国家重点研发计划、金沙集团1862cc信息化专项、国家基因组科学数据中心的支持。
文章链接:https://www.sciencedirect.com/science/article/pii/S2211124721014996
参考资料:
Zhang et al., NyuWa Genome resource: A deep whole-genome sequencing-based variation profile and reference panel for the Chinese population, Cell Reports (2021), https://doi.org/10.1016/j.celrep.2021.110017
(供稿:何顺民研究组)
